


Capstone believes Food and Drug Administration (FDA) guidance that adds new and overlapping compliance requirements is complicating the process of rapidly expanding clinical trials. As a result, clinical trial sponsors, such as pharmaceutical and biotech companies, will increasingly rely on end-to-end solutions to gain conviction in their data and processes, manage compliance burdens, and reduce the risk of costly failure.
- FDA guidance is raising the bar for compliance in clinical trials for new pharmaceuticals and medical devices. As federal regulations become more complex, sponsors face greater operational burdens, which drives demand for technology that simplifies workflow. In parallel, research and discovery (R&D) spending continues to rise as clinical trials spend stabilizes around pre-pandemic norms, benefiting end-to-end solutions.
- Access to the FDA for pre-submission feedback is more limited due to recent government workforce reductions. Given the cost of failed trials, there is strong demand for solutions that help sponsors build confidence in their submissions before filing.
- Sponsors and contract research organizations (CROs) cite fragmented tech stacks as a top pain point while recognizing that each point solution in the stack is necessary. This, in turn, creates an advantage for end-to-end platforms that consolidate systems while maintaining functionality.
Clinical Trials Landscape
Under the Trump administration, regulatory changes and evolving priorities across the clinical trials landscape—from development to post-market surveillance—have complicated the landscape for developers and sponsors aiming to bring drugs to market in the US.
The combination of new guidance, which creates more compliance burdens for sponsors, and FDA staffing reductions, which have caused administrative bottlenecks, leaves sponsors with fewer resources to use in an already demanding, high-pressure process. Constant guidance changes create a moving compliance target for sponsors that, without the direct access to FDA staff that they previously had, must find other ways to build conviction in the quality and integrity of the data in their final submission to the FDA. To accomplish this, sponsors will have to implement more technologies across the clinical trials lifecycle to manage compliance burdens and reduce the risk of costly failures.
Additionally, the clinical trials market is growing in dollars and is steady in volume, after re-adjusting from highs, indicative of a growing end-market for clinical trial technology (see Exhibit 1). Because drug development under artificial intelligence (AI) has shown early promise, but is not fully operational, Capstone expects growth in the total number of clinical trials by reducing the time and costs of drug development.
Exhibit 1: Total Clinical Trials Starts, K (RHS), and R&D Expenditure, $B (LHS)
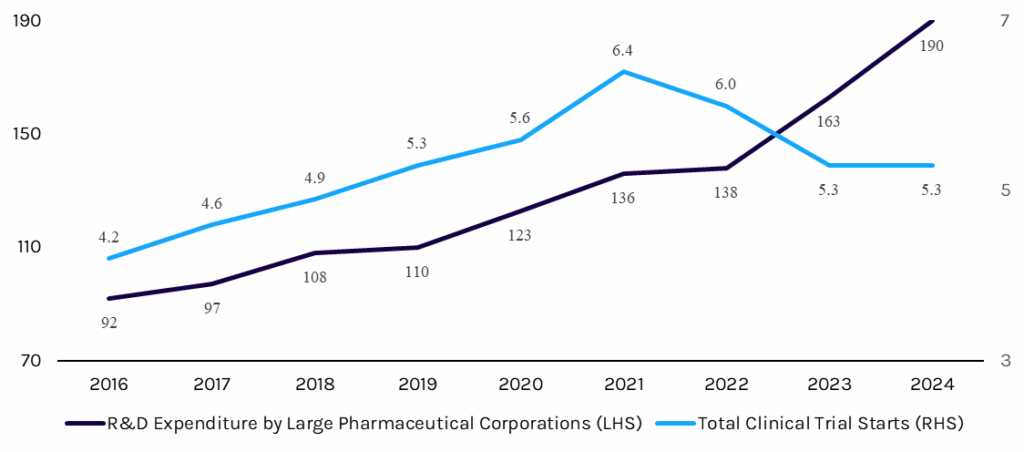
Source: IQVIA Global Trends in R&D 2025
Clinical Trials Overview
As a part of the FDA’s multi-stage process for pharmaceutical market authorization, a developer often must conduct clinical trials to establish the safety and efficacy of investigational products (IP). Clinical trials can be site-based, decentralized, or hybrid. Developing new pharmaceuticals and bringing them to market is an expensive, heavily regulated process. Sponsors often cannot afford to make significant errors or face regulatory setbacks, which creates demand for tools that can ensure data quality, compliance, and FDA submission readiness at each stage of the FDA approval process.
Clinical Trials Process
Drug development has three critical stages: preclinical research, clinical trials (Phases I-III), and post-approval studies (Phase IV). After a sponsor has conducted preclinical trial research and participated in preclinical trial consultations with the FDA and the Center for Drug Evaluation and Research (CDER), sponsors will submit an investigational new drug (IND) application and launch the clinical trials process.
Launch
Sponsors must contend with constantly evolving guidance—spanning from trial design to data standards to technology requirements—with a convoluted and overlapping set of compliance obligations. As the FDA prioritizes more rapid innovation, sponsors, or their CROs, will have to make critical investments in compliance-enabling technology.
A sponsor will begin by deciding whether it will be traditional and at a specific clinical trial site, hybrid, or fully decentralized clinical trial (DCT). Under the 2024 final DCT guidance, sponsors have directions from the FDA on how to conduct decentralized clinical trials, where participants can collect their own data and outcomes in their home, as well as how a local practitioner, who is not a trial administrator, can collect their data.
Decentralized Trials
DCTs allow some or all trial-related activities to take place at locations other than traditional DCT sites, including at the homes of trial participants or at local healthcare facilities. Sponsors that conduct DCTs must ensure that protocols are being properly followed and that the data are accurate. As a result, the FDA released the DCT guidance that included a new compliance regime that required sponsors to maintain and submit data to verify the accuracy of their findings. Sponsors must complete a documented data management plan that includes all the methodologies and technologies used for remote data collection, a list of all service providers interacting with data, study records that capture all specifics of a visit, a trial-monitoring plan to assess protocol compliance and data integrity, and compliance records on privacy rules. In the final guidance, the FDA calls out that sponsors may wish to adopt technological solutions, including an Electronic Case Report Form (eCRF), to help manage the data for compliance burden; the FDA had previously finalized guidance for these digital health technologies (DHTs) in 2023.
Sponsors participating in DCTs can have participants collect their own data with a wearable technology that tracks biometrics or an Electronic Clinical Outcome Assessment (eCOA) or Electronic Patient-Reported Outcomes (ePRO) platforms. These tools give sponsors more confidence in their results, as they enable regular monitoring of participants, regardless of physical proximity.
Real-World Data
Sponsors then determine what data they intend to collect and the external data they need. Device manufacturers, since December 2025, have been allowed to use real-world data (RWD) as evidence in their clinical trials without connecting each data point to an identifiable patient record. The FDA has indicated that it intends to allow drug clinical trials to use RWD as clinical evidence in the near future.
This change will allow sponsors to more easily leverage large, aggregated datasets, such as electronic health records (EHR), commercial claims, and product and disease registries, as clinical evidence. We believe this will create opportunities for sponsors by allowing them to construct external controls for rare disease trials, conduct post-market surveillance, support label expansions, and reduce time to market without collecting the data themselves.
However, to use RWD, sponsors must record data provenance, collection methods, and quality controls, and must verify the reliability and relevance of the data to their trial‒creating a new compliance burden. RWD is not intended for clinical trial use and often requires cleaning or manipulation to be fit-for-purpose. As sponsors implement RWD in device (and, eventually, drug) clinical trials, technology that helps prepare and analyze data, including analytics and visualization platforms, will become essential to operations and analysis.
Trial Implementation
After sponsors have submitted their IND application, they can launch their trials. Phase I of a clinical trial evaluates safety, both Phases II and III evaluate efficacy, and after Phase III, sponsors submit a new drug application (NDA) for small molecule drugs or a biologics license application (BLA) for biological products. The FDA will either approve the applications or sponsors will receive a complete response letter (CRL), notifying them that their application is not ready for approval and must be resubmitted with additional data. Once approved, the drug moves into Phase IV, which is post-market surveillance.
During the clinical trials process, regardless of trial design, sponsors or CROs use tools to manage the clinical trial, to collect data, and to ensure continuing protocol and regulatory compliance (e.g., Electronic Data Capture (EDC) tools allow a clinician to record structured data about a trial participant and Clinical Trial Management Systems (CTMS) house study protocols, site monitoring and patient recruitment tools, and can assist with regulatory compliance). Overall, the operational burden of managing the trial‒especially large clinical Phase III trials, which can span multiple states and dozens of sites‒can be a significant task on its own, which clinical trials tech can manage.
Guidance Gap
Sponsors generally take part in pre-submission FDA meetings, as well as regular back-and-forth with the agency, throughout the clinical trial process. However, in March 2025, the Department of Government Efficiency announced federal workforce cuts. For the FDA, these reductions in force focused on administrative functions and exempted drug and device review staff.
While the FDA continues to meet their Prescription Drug User Fee Act (PDUFA) target dates for application review, industry reports suggest that the wait for a pre-IND meeting had – as of May 2025 – had increased from three to six months in some cases and that sponsors should expect fewer opportunities to engage directly with the FDA. Additionally, some parts of the FDA have turned to replacing in-person meetings with written feedback.
Historically, these meetings with the FDA have allowed sponsors to tailor their submission, as they develop it, to the FDA’s expectations and feedback. If sponsors cannot receive regular feedback from or interact with the FDA, then it will be critical for them to leverage technologies to ensure their data is complete and compliant to gain approval. As such, we believe sponsors increase reliance on clinical trial technologies that can provide some assurance of the quality of their submission to the FDA.
Implications for Investment
Staff on clinical trial investigative sites recognize the value of clinical trial technology solutions. More than 93% report that they believe remote patient monitoring (RPM) or management solutions are essential. In addition, most sites said they have invested in at least one clinical trial digital tool. Among the most adopted technologies are digital solutions, such as eDiaries/ePRO/eCOA, which allow patients to report their outcomes, as well as solutions that facilitate interactions between trial administrators and participants, such as telehealth (see Exhibit 2).
Exhibit 2: Site Investments in Reporting Tools, 2025
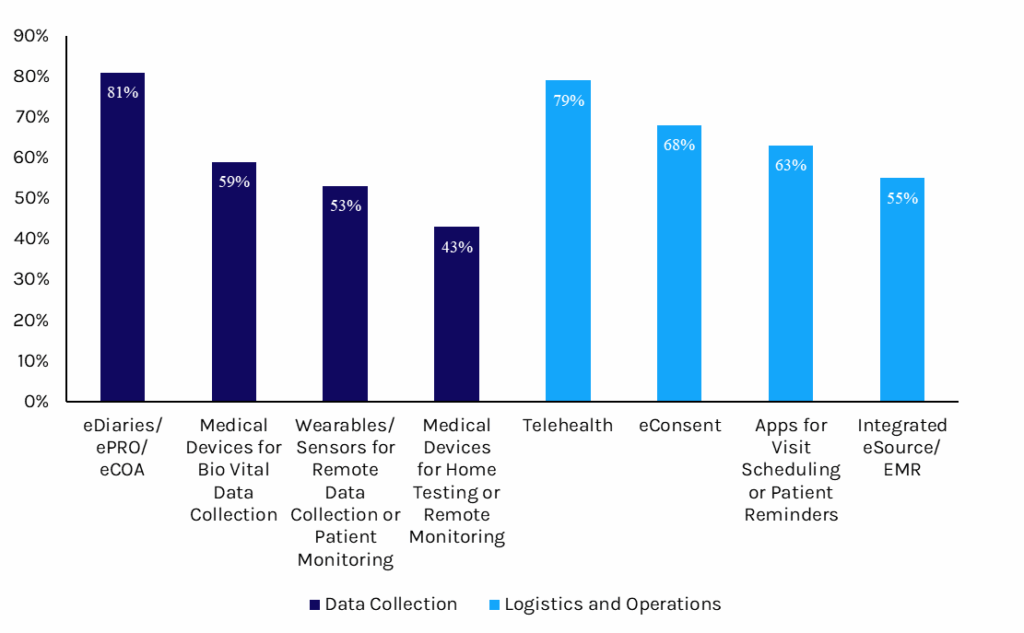
Source: Tufts CSDD
Clinical trial operators and administrators report in industry surveys that the biggest pain point with their digital solutions is coordination across solutions. Nearly all survey respondents note that better integration between data and operating systems would improve trial execution. CROs and sponsors also highlighted an inherent contradiction in clinical trial technology implementation: it is both a priority to invest in technologies to create efficiencies and improve data quality, but sites are already burdened with too many technologies that are not interoperable and do not enable efficient data sharing between platforms.
This paradox creates an opportunity for investment in clinical trial technology. The current market fragmentation can be solved by coordinated solutions that cover the entire clinical trial lifecycle, rather than the disjointed implementation of point solutions. Strategies that focus on acquiring end-to-end solutions or the acquisition of multiple point solutions to consolidate into an end-to-end solution are likely to see the greatest success in this market.
Artificial Intelligence Investment
Forecasts of the impacts of artificial intelligence on drug development suggest that agentic AI could increase productivity in clinical development 35%-45% during the next five years, creating opportunities for clinical trial technology as the end market expands. However, as AI supports the automation of the clinical trial process—for example, with site selection and coordination—the risk of disintermediation of digital solutions increases. Solutions that effectively leverage AI to improve their offerings, by adding AI support for data monitoring, analysis, and validation, as well as to increase efficiency in their operational offerings, will be positioned to best capture the increased volume created by AI. Capstone also notes that vendors that solve the pain points for FDA-mandated evidentiary standards, such as tracking data flow or provenance and fit-for-purpose validation, will be more difficult to disintermediate, as they create a record that is integral to an FDA application submission, rather than solely creating operational efficiencies that could be replaced by an AI agent.
Read more from Capstone’s healthcare team:
The ABA Medicaid Rate Reset
What Will Vaccine Policy Chaos Cost Payors?
European Digitization Gaps Create Opportunities for Healthcare IT Investment




